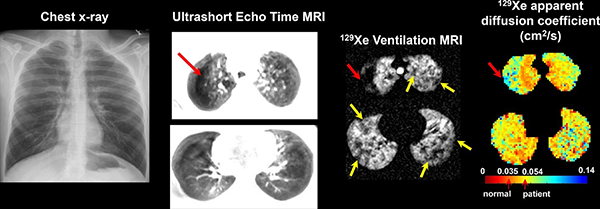
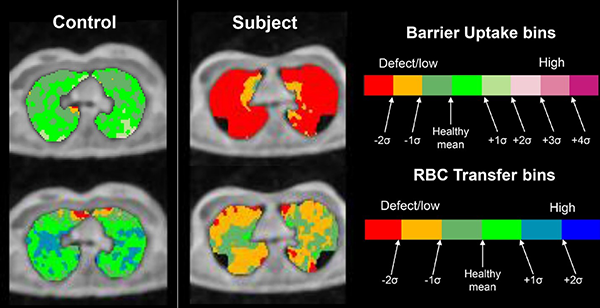
Obstructive Lung Diseases
Cystic Fibrosis
Cystic fibrosis (CF) is a lifelong disease that affects the respiratory, endocrine, reproductive, and digestive systems. About 30,000 children and adults in the United States (70,000 worldwide) have CF. This disease is caused by a defective gene that makes the body produce very thick, sticky mucus.
The cystic fibrosis transmembrane conductance regulator (CFTR) protein helps to maintain the balance of salt and water on many surfaces in the body, such as the surface of the lung. When the protein is not working correctly, chloride (a component of salt) becomes trapped in cells. Without the proper movement of chloride, water cannot hydrate the cellular surface. This leads the mucus covering the cells to become thick and sticky, causing many of the symptoms associated with cystic fibrosis.
We hypothesize that MRI will serve as a sensitive tool to longitudinally monitor CF lung disease progression and response to CFTR modulator therapy in children with CF, without exposure to ionizing radiation. We use advances in MRI technology to longitudinally evaluate CF lung disease via MRI, assessing disease progression and therapeutic efficacy in young CF patients precisely when CFTR modulators enter the clinic.
We do this with the following aims:
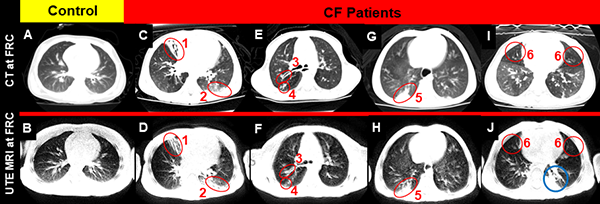
- To detect, quantify, and validate regional lung disease by UTE MRI through comparison with CT and pulmonary function tests in pediatric CF patients.
- To use UTE MRI to longitudinally quantify differences in disease trajectory between CF children undergoing CFTR modulator therapy and CF children with two CFTR mutations who are ineligible for therapy or are not treated with a CFTR modulator.
- To examine relationships between cross-sectional and longitudinal changes in UTE MRI and compare with sensitive tools that monitor CF ventilation, including hyperpolarized 129Xe ventilation MRI and lung clearance index (LCI).